
Have you ever wondered if your generic medication is actually as effective as the brand-name version? It’s a question that haunts many patients and even some healthcare professionals. The short answer is yes, but the math behind that assurance is surprisingly specific. Regulatory agencies worldwide rely on a standard known as the 80-125% rule, which serves as the global benchmark for determining bioequivalence between pharmaceutical products. This rule doesn’t mean generics contain 80% to 125% of the active ingredient-that’s a common myth. Instead, it governs how quickly and completely your body absorbs the drug.
To understand why this range matters, we need to look at what happens when you swallow a pill. Your body doesn’t just absorb the drug instantly; it processes it over time. Scientists measure this process using two key metrics: Area Under the Curve (AUC), representing total drug exposure in the bloodstream over time, and Cmax, representing the maximum concentration of the drug reached in the blood. If these values for a generic drug fall within the 80-125% confidence interval of the brand-name drug, they are considered bioequivalent. This means they will perform similarly in your body.
The Myth of the "Weaker" Generic
Let’s clear up the biggest misconception right away. Many people believe the 80-125% rule allows manufacturers to cut corners, putting less medicine into generic pills. In reality, the amount of active ingredient in both generic and brand drugs must be identical, typically within a tight 95-105% tolerance of the label claim. The 80-125% rule applies to pharmacokinetics-how your body handles the drug-not the formulation itself.
Think of it like two different cars from the same manufacturer. They might have slightly different paint or interior materials (excipients), but their engines (active ingredients) are built to the same specifications. The 80-125% rule ensures that both cars accelerate and handle the road in essentially the same way. A 2022 survey by the American Pharmacists Association found that 63% of community pharmacists mistakenly believed the rule referred to the quantity of active ingredient, highlighting how pervasive this misunderstanding is.
Why 80-125% and Not ±20%?
You might ask why the range isn’t simply -20% to +20%. The answer lies in statistics and biology. Drug absorption data usually follows a log-normal distribution, meaning it’s skewed rather than perfectly symmetrical. To analyze this data accurately, scientists use logarithmic transformation. On this transformed scale, the range becomes symmetric around zero, specifically from -0.2231 to +0.2231. When you convert those numbers back to the original scale, they become 80% and 125%.
This mathematical adjustment is crucial because it accounts for the proportional nature of biological variability. A difference of 20% in absorption is clinically insignificant for most drugs, as established during the pivotal 1986 FDA Bioequivalence Hearing. Experts concluded that such minor variations would not affect patient outcomes. By using geometric means and confidence intervals, regulators ensure that the average performance of the generic matches the reference product closely enough to be safe and effective.
| Misconception | Scientific Reality |
|---|---|
| Generics contain 80-125% of the active ingredient | Generics contain 95-105% of the labeled dose, identical to brands |
| The rule uses a 95% confidence interval | The rule strictly requires a 90% confidence interval |
| Any drug can vary by 25% | Narrow therapeutic index drugs often require tighter limits (90-111%) |
| Bioequivalence tests clinical efficacy directly | Bioequivalence tests absorption rates (AUC and Cmax) in healthy volunteers |
The Role of the 90% Confidence Interval
Another point of confusion is the use of a 90% confidence interval instead of the more familiar 95%. In statistical terms, a 90% confidence interval means we are 90% sure that the true population mean falls within the calculated range. For bioequivalence, this allows for a 5% error rate at both the upper and lower bounds, totaling a 10% risk. This is stricter than traditional hypothesis testing, which could fail to detect small but meaningful differences if the sample size was too large.
The requirement is that the entire 90% confidence interval of the ratio of geometric means for both AUC and Cmax must fall within the 80-125% window. If even a tiny part of the interval crosses outside this boundary, the generic fails. This rigorous standard ensures that while individual patients may experience slight variations, the overall population will see consistent results. The FDA mandates that both the point estimate and the confidence interval meet this criterion, adding an extra layer of safety.
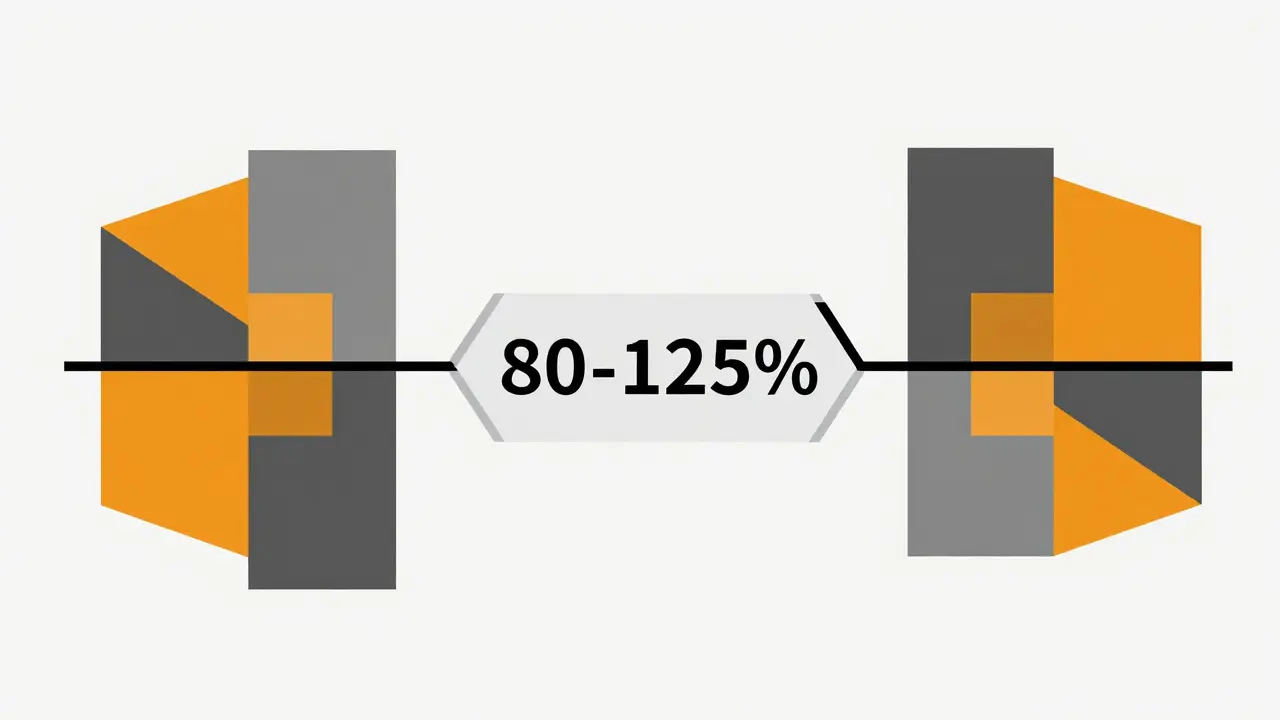
Exceptions: Narrow Therapeutic Index Drugs
While the 80-125% rule works well for most medications, it’s not a one-size-fits-all solution for every drug. Some medications, known as Narrow Therapeutic Index (NTI) drugs, are medications where small changes in dosage can lead to serious side effects or loss of efficacy, require much tighter control. Examples include warfarin, levothyroxine, and certain anti-seizure medications. For these drugs, a 20% difference in absorption could be dangerous.
Regulatory agencies like the FDA and EMA often apply stricter limits for NTI drugs, such as a 90-111% confidence interval. This reflects the higher stakes involved. Dr. Lawrence Lesko, former Director of the Office of Clinical Pharmacology at the FDA, noted that while the 80-125% criterion was based on clinical judgment, it has proven robust for most drugs. However, for NTI drugs, the margin for error is simply too small to allow the standard range. Patients taking these medications should always consult their doctors before switching manufacturers, although modern standards make this increasingly safe.
High-Variability Drugs and Scaled Approaches
On the other end of the spectrum are high-variability drugs. These are medications where natural human variation in absorption is very high, often with a coefficient of variation greater than 30%. For these drugs, meeting the standard 80-125% rule can be statistically difficult, even if the drug is perfectly equivalent. To address this, regulators have introduced Scaled Average Bioequivalence (SABE), a statistical method that widens the acceptance range for highly variable drugs based on the reference product's variability.
Under SABE, the acceptance range can expand up to 69.84-143.19% for Cmax, depending on how variable the reference drug is. This approach acknowledges that if the original drug varies widely from person to person, a generic doesn’t need to be artificially precise beyond that natural variability. This method is used by the European Medicines Agency and is under consideration by the FDA for specific cases, ensuring that useful generics aren’t rejected due to statistical quirks rather than actual performance issues.

Global Harmonization and Market Impact
The adoption of the 80-125% rule has been a game-changer for the global pharmaceutical industry. Before this standard, countries had different criteria, making international drug development costly and complex. Today, major health authorities including the FDA, EMA, WHO, and Health Canada use nearly identical standards. This harmonization, driven by groups like the International Council for Harmonisation (ICH), has accelerated the approval of generic drugs worldwide.
The impact is significant. Since the Hatch-Waxman Act of 1984, over 14,000 generic drugs have been approved in the U.S. alone. Generics now account for 90% of all prescriptions filled in the United States but only 23% of total drug spending. This cost savings is largely due to the streamlined bioequivalence process, which avoids the need for expensive clinical efficacy trials. Instead, companies focus resources on proving that their product behaves like the reference drug in the body, saving billions in healthcare costs annually.
Future Directions: Personalized Bioequivalence
As science advances, so does our understanding of how drugs interact with the body. Emerging research in pharmacogenomics suggests that future bioequivalence standards may need to account for genetic variations in drug metabolism. Currently, the 80-125% rule treats all patients as a single population. However, individuals with different genetic profiles may metabolize drugs differently, potentially requiring personalized bioequivalence criteria by 2030.
The FDA’s 2023-2027 Strategic Plan includes modernizing bioequivalence tools, with funding allocated for model-informed approaches. Additionally, the Complex Generics Initiative addresses challenges with novel drug delivery systems like inhalers and topical products, where traditional oral bioequivalence metrics don’t apply. While the 80-125% rule remains the gold standard for immediate-release solid oral dosage forms, the landscape is evolving to meet the complexities of modern medicine.
Does the 80-125% rule mean generics are weaker?
No. The rule refers to the confidence interval of absorption rates (AUC and Cmax), not the amount of active ingredient. Generics must contain the same dose as brand-name drugs, typically within a 95-105% range.
Why is a 90% confidence interval used instead of 95%?
A 90% confidence interval allows for a 5% error rate at each limit, totaling 10%. This is statistically rigorous for equivalence testing and prevents false rejections of bioequivalent products due to overly strict margins.
What are narrow therapeutic index drugs?
These are drugs where small changes in blood levels can cause serious side effects or treatment failure. Examples include warfarin and levothyroxine. They often require tighter bioequivalence limits, such as 90-111%.
How do bioequivalence studies work?
Studies typically involve 24-36 healthy volunteers in a crossover design. Participants take both the test and reference drugs, and blood samples are analyzed to calculate AUC and Cmax. The geometric mean ratios must fall within the 80-125% range.
Is the 80-125% rule used globally?
Yes. Major regulatory bodies including the FDA, EMA, WHO, and Health Canada use this standard. This harmonization facilitates international drug approval and reduces development costs for generic manufacturers.













